
Редкий диагноз СМА (спинальная мышечная атрофия) у многих на слуху, но до сих пор не для всех очевидно, из-за чего возникает это заболевание и какие есть перспективы в лечении. В месяц осведомленности о СМА собрали для вас основную информацию.
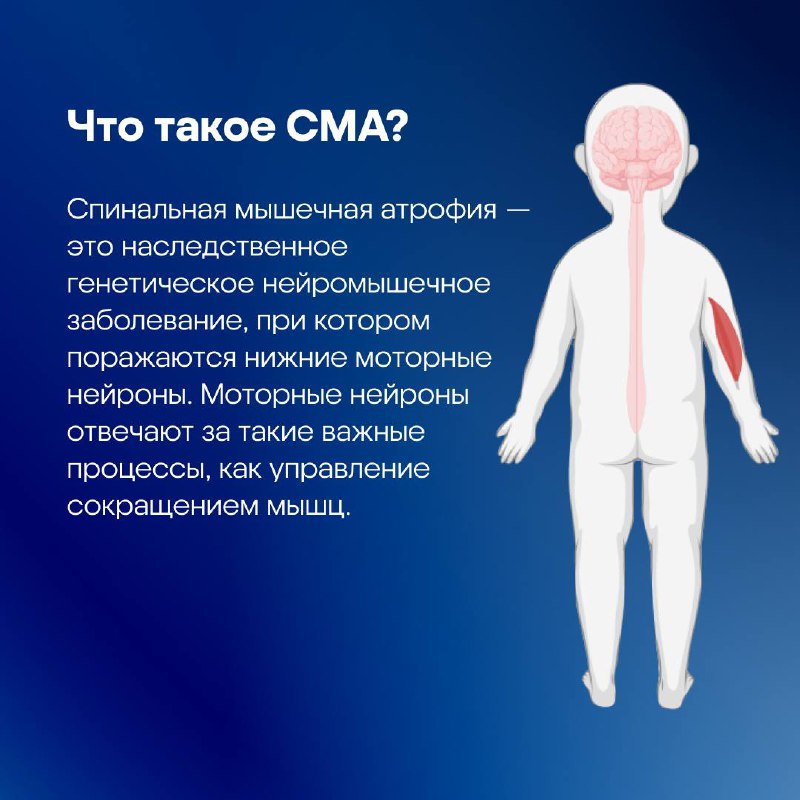
СМА — это наследственное генетическое нейромышечное заболевание, при котором поражаются нижние моторные нейроны — клетки нервной системы, отвечающие за движение. Что это значит?
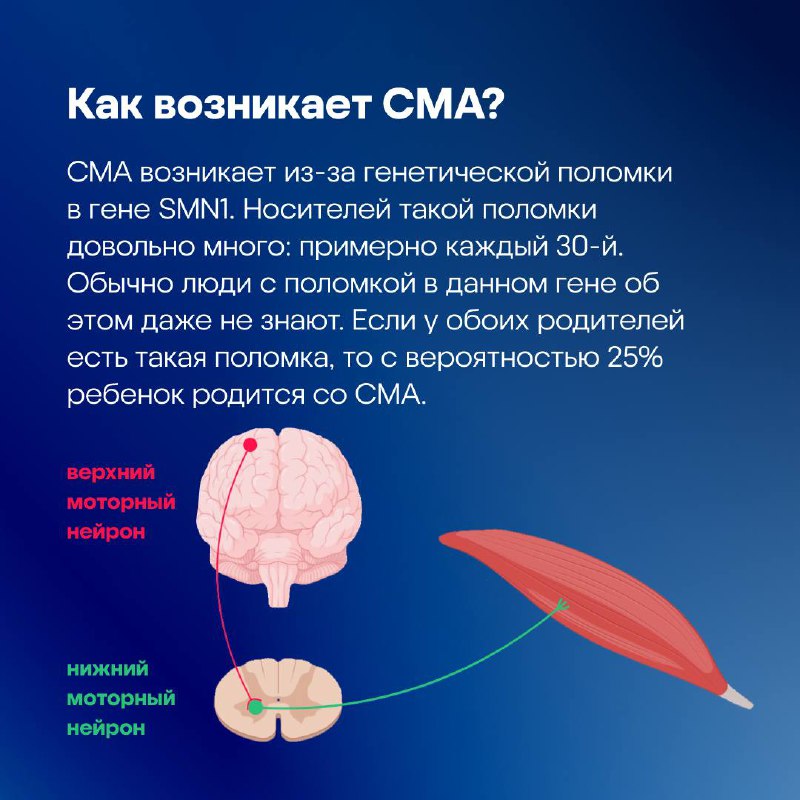
Когда вы хотите сократить какую-либо мышцу, то первой в дело вступает центральная нервная система. Верхние моторные нейроны головного мозга отправляют сигнал в спинной мозг, где его принимают нижние моторные нейроны, управляющие сокращением мышц по всему организму: от движения руками до, например, дыхания. В случае если нижний моторный нейрон по какой-то причине погибает, сигналы к мышцам перестанут проходить, несмотря на то что головной мозг продолжит их посылать. Это и происходит при СМА.
Причина СМА — недостаток всего одного белка выживания моторных нейронов (SMN). Каждый человек имеет две копии гена SMN1, кодирующего этот белок, — по одной на каждой из двух версий 5-й хромосомы: от матери и от отца. Если у обоих родителей только по одной рабочей копии гена SMN1, то у ребенка есть 25% вероятность рождения без единого корректного гена SMN1, что и приводит к возникновению СМА.
Эволюция позаботилась о «резервной копии» такого важного механизма, а потому у людей также есть ген SMN2, который также кодирует тот же белок SMN, но гораздо менее эффективно — большая часть получающихся белков нефункциональна из-за точечной мутации. Тем не менее работа гена SMN2 может частично компенсировать отсутствие гена SMN1, хоть его эффективности и недостаточно для полноценного функционирования моторных нейронов. В случае поломки SMN1 именно количество количества копий гена SMN2 будет определять тяжесть поражения моторных нейронов. В совокупности с возрастом начала заболевания и степенью нарушений этот фактор определяет тип СМА: I, II, III и IV. Самыми тяжелыми считаются типы I и II, при которых у людей наблюдаются значительные ограничения в движениях, нарушения дыхания и глотания.
С учетом патогенеза заболевания есть два основных подхода к терапии:
1. Стимуляция гена SMN2, чтобы увеличить количественно и качественно выработку белка SMN, который помогает выживать моторным нейронам.
2. Доставка корректной копии гена SMN1, что может быть осуществлено с помощью генной терапии. Это может привести к выработке полноценной и функциональной версии белка SMN.
Сейчас наша команда работает над препаратом, в основе которого лежит генная терапия с помощью вирусной доставки генов. Препарат для терапии СМА I типа находится на стадии клинических исследований. По мере получения информации от наших ученых мы будем сообщать вам о результатах в наших соцсетях.
СМА — это наследственное генетическое нейромышечное заболевание, при котором поражаются нижние моторные нейроны — клетки нервной системы, отвечающие за движение. Что это значит?
Когда вы хотите сократить какую-либо мышцу, то первой в дело вступает центральная нервная система. Верхние моторные нейроны головного мозга отправляют сигнал в спинной мозг, где его принимают нижние моторные нейроны, управляющие сокращением мышц по всему организму: от движения руками до, например, дыхания. В случае если нижний моторный нейрон по какой-то причине погибает, сигналы к мышцам перестанут проходить, несмотря на то что головной мозг продолжит их посылать. Это и происходит при СМА.
Причина СМА — недостаток всего одного белка выживания моторных нейронов (SMN). Каждый человек имеет две копии гена SMN1, кодирующего этот белок, — по одной на каждой из двух версий 5-й хромосомы: от матери и от отца. Если у обоих родителей только по одной рабочей копии гена SMN1, то у ребенка есть 25% вероятность рождения без единого корректного гена SMN1, что и приводит к возникновению СМА.
Эволюция позаботилась о «резервной копии» такого важного механизма, а потому у людей также есть ген SMN2, который также кодирует тот же белок SMN, но гораздо менее эффективно — большая часть получающихся белков нефункциональна из-за точечной мутации. Тем не менее работа гена SMN2 может частично компенсировать отсутствие гена SMN1, хоть его эффективности и недостаточно для полноценного функционирования моторных нейронов. В случае поломки SMN1 именно количество количества копий гена SMN2 будет определять тяжесть поражения моторных нейронов. В совокупности с возрастом начала заболевания и степенью нарушений этот фактор определяет тип СМА: I, II, III и IV. Самыми тяжелыми считаются типы I и II, при которых у людей наблюдаются значительные ограничения в движениях, нарушения дыхания и глотания.
С учетом патогенеза заболевания есть два основных подхода к терапии:
1. Стимуляция гена SMN2, чтобы увеличить количественно и качественно выработку белка SMN, который помогает выживать моторным нейронам.
2. Доставка корректной копии гена SMN1, что может быть осуществлено с помощью генной терапии. Это может привести к выработке полноценной и функциональной версии белка SMN.
Сейчас наша команда работает над препаратом, в основе которого лежит генная терапия с помощью вирусной доставки генов. Препарат для терапии СМА I типа находится на стадии клинических исследований. По мере получения информации от наших ученых мы будем сообщать вам о результатах в наших соцсетях.
tg-me.com/BIOCAD/440
Create:
Last Update:
Last Update:
Редкий диагноз СМА (спинальная мышечная атрофия) у многих на слуху, но до сих пор не для всех очевидно, из-за чего возникает это заболевание и какие есть перспективы в лечении. В месяц осведомленности о СМА собрали для вас основную информацию.
СМА — это наследственное генетическое нейромышечное заболевание, при котором поражаются нижние моторные нейроны — клетки нервной системы, отвечающие за движение. Что это значит?
Когда вы хотите сократить какую-либо мышцу, то первой в дело вступает центральная нервная система. Верхние моторные нейроны головного мозга отправляют сигнал в спинной мозг, где его принимают нижние моторные нейроны, управляющие сокращением мышц по всему организму: от движения руками до, например, дыхания. В случае если нижний моторный нейрон по какой-то причине погибает, сигналы к мышцам перестанут проходить, несмотря на то что головной мозг продолжит их посылать. Это и происходит при СМА.
Причина СМА — недостаток всего одного белка выживания моторных нейронов (SMN). Каждый человек имеет две копии гена SMN1, кодирующего этот белок, — по одной на каждой из двух версий 5-й хромосомы: от матери и от отца. Если у обоих родителей только по одной рабочей копии гена SMN1, то у ребенка есть 25% вероятность рождения без единого корректного гена SMN1, что и приводит к возникновению СМА.
Эволюция позаботилась о «резервной копии» такого важного механизма, а потому у людей также есть ген SMN2, который также кодирует тот же белок SMN, но гораздо менее эффективно — большая часть получающихся белков нефункциональна из-за точечной мутации. Тем не менее работа гена SMN2 может частично компенсировать отсутствие гена SMN1, хоть его эффективности и недостаточно для полноценного функционирования моторных нейронов. В случае поломки SMN1 именно количество количества копий гена SMN2 будет определять тяжесть поражения моторных нейронов. В совокупности с возрастом начала заболевания и степенью нарушений этот фактор определяет тип СМА: I, II, III и IV. Самыми тяжелыми считаются типы I и II, при которых у людей наблюдаются значительные ограничения в движениях, нарушения дыхания и глотания.
С учетом патогенеза заболевания есть два основных подхода к терапии:
1. Стимуляция гена SMN2, чтобы увеличить количественно и качественно выработку белка SMN, который помогает выживать моторным нейронам.
2. Доставка корректной копии гена SMN1, что может быть осуществлено с помощью генной терапии. Это может привести к выработке полноценной и функциональной версии белка SMN.
Сейчас наша команда работает над препаратом, в основе которого лежит генная терапия с помощью вирусной доставки генов. Препарат для терапии СМА I типа находится на стадии клинических исследований. По мере получения информации от наших ученых мы будем сообщать вам о результатах в наших соцсетях.
СМА — это наследственное генетическое нейромышечное заболевание, при котором поражаются нижние моторные нейроны — клетки нервной системы, отвечающие за движение. Что это значит?
Когда вы хотите сократить какую-либо мышцу, то первой в дело вступает центральная нервная система. Верхние моторные нейроны головного мозга отправляют сигнал в спинной мозг, где его принимают нижние моторные нейроны, управляющие сокращением мышц по всему организму: от движения руками до, например, дыхания. В случае если нижний моторный нейрон по какой-то причине погибает, сигналы к мышцам перестанут проходить, несмотря на то что головной мозг продолжит их посылать. Это и происходит при СМА.
Причина СМА — недостаток всего одного белка выживания моторных нейронов (SMN). Каждый человек имеет две копии гена SMN1, кодирующего этот белок, — по одной на каждой из двух версий 5-й хромосомы: от матери и от отца. Если у обоих родителей только по одной рабочей копии гена SMN1, то у ребенка есть 25% вероятность рождения без единого корректного гена SMN1, что и приводит к возникновению СМА.
Эволюция позаботилась о «резервной копии» такого важного механизма, а потому у людей также есть ген SMN2, который также кодирует тот же белок SMN, но гораздо менее эффективно — большая часть получающихся белков нефункциональна из-за точечной мутации. Тем не менее работа гена SMN2 может частично компенсировать отсутствие гена SMN1, хоть его эффективности и недостаточно для полноценного функционирования моторных нейронов. В случае поломки SMN1 именно количество количества копий гена SMN2 будет определять тяжесть поражения моторных нейронов. В совокупности с возрастом начала заболевания и степенью нарушений этот фактор определяет тип СМА: I, II, III и IV. Самыми тяжелыми считаются типы I и II, при которых у людей наблюдаются значительные ограничения в движениях, нарушения дыхания и глотания.
С учетом патогенеза заболевания есть два основных подхода к терапии:
1. Стимуляция гена SMN2, чтобы увеличить количественно и качественно выработку белка SMN, который помогает выживать моторным нейронам.
2. Доставка корректной копии гена SMN1, что может быть осуществлено с помощью генной терапии. Это может привести к выработке полноценной и функциональной версии белка SMN.
Сейчас наша команда работает над препаратом, в основе которого лежит генная терапия с помощью вирусной доставки генов. Препарат для терапии СМА I типа находится на стадии клинических исследований. По мере получения информации от наших ученых мы будем сообщать вам о результатах в наших соцсетях.
BY BIOCAD


Share with your friend now:
tg-me.com/BIOCAD/440